




2022年11月15日,华东医药发布公告称,美国合作方ImmunoGen,Inc.对外宣布,其用于治疗铂耐药卵巢癌的全球首创(first-in-class)ADC药物ELAHERE™(mirvetuximab soravtansine-gynx,研发代码:IMGN853、HDM2002)获得美国食品药品监督管理局(FDA)加速批准上市。这也是全球第15款获批上市的ADC药物。

截图来源:企业公告
全球获批上市的15款ADC药物大盘点
抗体药物偶联物(antibody drug conjugate),即ADC药物的设计理念由来已久,早在1913年,化疗之父、诺贝尔奖得主Paul Ehrlich教授首次提出“魔力子弹(magic bullet)”的概念,即将细胞毒性药物安装在特异性单克隆抗体上,实现定向杀伤肿瘤细胞。
目前,全球已有15款ADC药物获批上市,分别是辉瑞的Mylotarg、Besponsa,罗氏的Kadcyla、Polivy、阿斯利康的Lumoxiti、Enhertu,Seagen/武田制药的Adcetris、Padcev,Seagen/Genmab的Tivdak,葛兰素史克的Blenrep、吉利德的Trodelvy、Rakuten Medical的Akalux,ADCTherapeutics的Zynlonta、荣昌生物的维迪西妥单抗,以及此次ImmunoGen/华东医药的Elahere,治疗疾病涉及淋巴瘤、白血病、乳腺癌、多发性骨髓瘤、乳腺癌、头颈癌、尿路上皮癌等。
其中,中国市场获批的ADC药物有5个,分别是罗氏的恩美曲妥珠单抗、Seagen/武田的维布妥昔单抗、辉瑞的奥加伊妥珠单抗、荣昌生物的维迪西妥单抗和Immunomedics的戈沙妥珠单抗。
全球获批上市的15款ADC药物大盘点
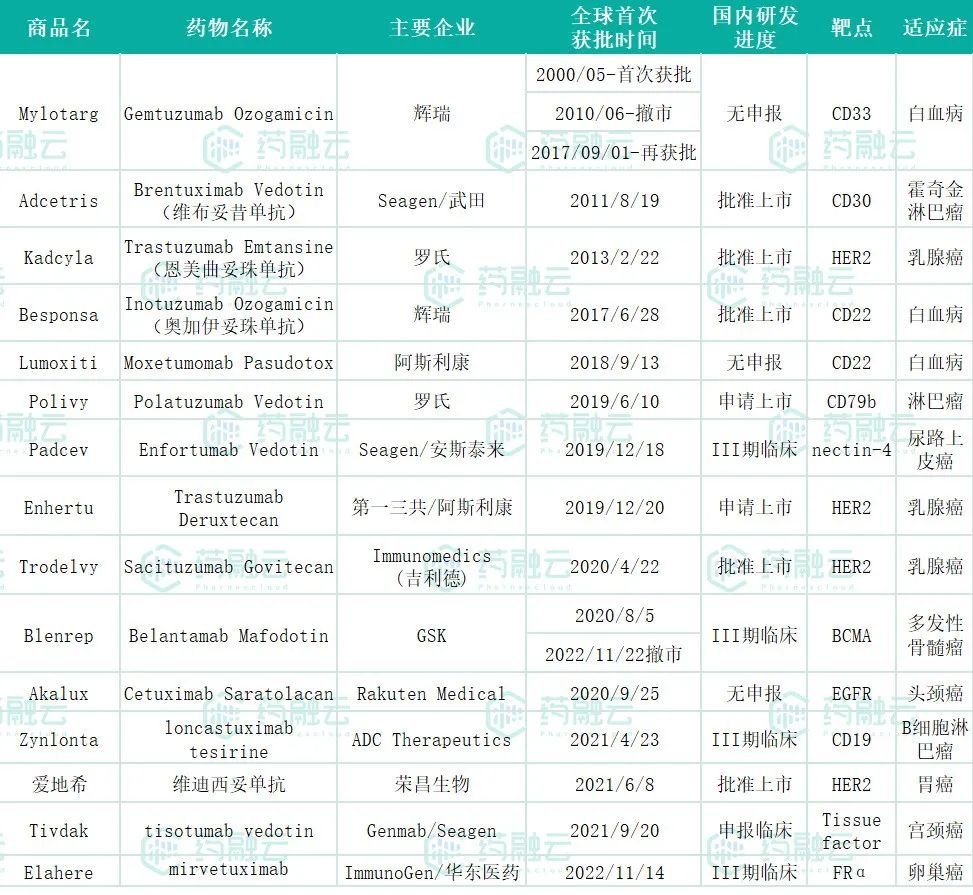
(数据来源:药融云全球药物研发、全球上市批文)
01.Gemtuzumab Ozogamicin(Mylotarg®:Pfizer/Wyeth)
Mylotarg(Gemtuzumab Ozogamicin)由辉瑞研发,是全球第一款上市的ADC药物。但上市后不久,因安全性问题于2010年6月宣布Mylotarg自主撤市。随后更新了临床证据,调整了规格,最终在2017年,Mylotarg通过将原来9mg/m2调整至3mg/m2,这款历经17年波折的ADC药物再次获得FDA审批上市。FDA批准该药物用于治疗2岁及以上的CD33阳性AML患者,这些患者经历复发或对初始治疗没有响应。
值得一提的是,Mylotarg是首款包括儿童AML适应症的药物,也是目前唯一一款靶向CD33的AML治疗方法。
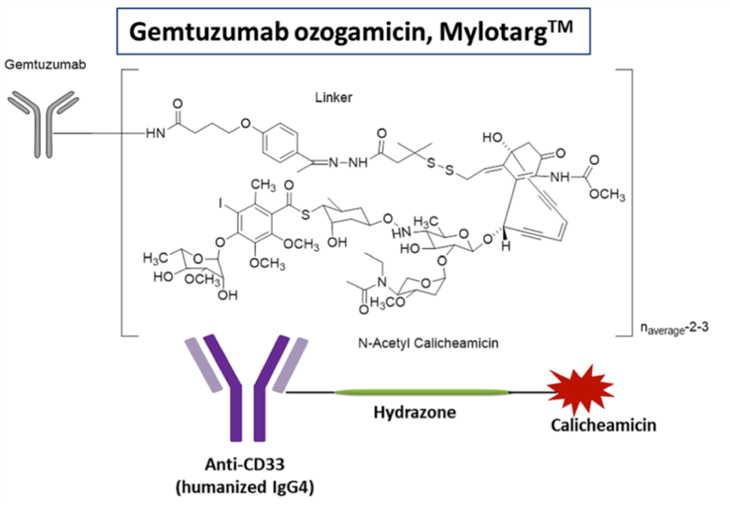
(Mylotarg®结构和生物特性,来源:Pifzer)
在2010年撤市之后,辉瑞进行了更充分的临床试验,补充了研究数据,第二次批准基于以下三项临床试验研究:ALFA-0701、AML-19、MyloFrance-1。
ALFA-0701是一项多中心、随机、开放标签的临床3期试验,共纳入271名CD33阳性的、新确诊的AML成人患者,患者随机分配到Mylotarg®联合治疗组和单用化疗组,主要终点是无事件生存期(EFS)。结果显示,Mylotarg®联合治疗组的EFS为17.3个月,而单用化疗组EFS为9.5个月。
AML-19是一项多中心、随机、开放标签的临床3期试验,对比单药Mylotarg®治疗与最佳治疗支持(BSC)的效果差异,研究在不能耐受其他AML治疗方法的、60岁以上的老年患者中进行。研究显示,接受Mylotarg®治疗患者的中位总生存期(OS)为4.9个月,而接受BSC的患者的中位OS为3.6个月。
MyloFrance-1是一项单臂、开放标签的临床2期试验,研究在57例首次复发的成年患者中展开。临床试验显示,15例患者达到完全缓解(CR),中位无复发生存期(RFS)为11.6个月。
02.Brentuximab Vedotin(Adcetris®:Seattlegenetics)
Adcetris®(brentuximab vedotin,维布妥昔单抗,安适利®),由Seagen(原Seattle Genetics)原研,后来与Takeda联合开发。2011年8月19日,维布妥昔单抗获得美国食品和药品监督管理局(FDA)首次批准,用于治疗霍奇金淋巴瘤和系统性间变性大细胞淋巴瘤。
2020年5月12日,获得我国国家药品监督管理局(NMPA)批准,正式进入中国,是第二款国内获批上市的ADC药物。2021年10月25日,获得欧盟批准,可作为CD30表达的系统性间变性大细胞淋巴瘤一线治疗药物,也成为全球第一个用于一线治疗的ADC药物。2021年Adcetris®全球销售额13.06亿美元,成为名副其实的重磅炸弹药物。
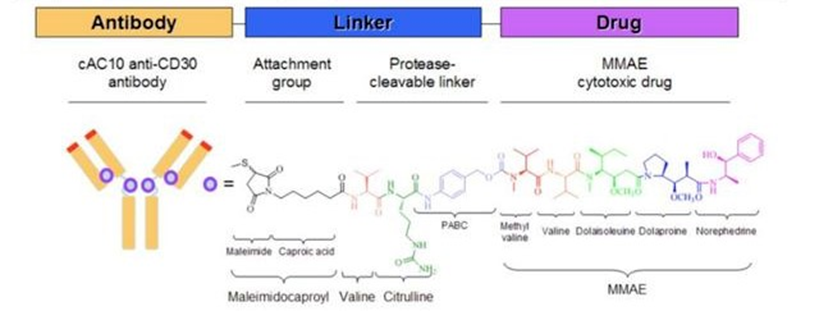
(Adcetris®结构,来源:Seagen)
Adcetris®由靶向CD30的单抗brentuximab和小分子毒性药物MMAE,通过二肽连接子偶联而成,属于第二代ADC代表药物,比第一代ADC更具优势。2011年FDA基于以下两项临床试验研究,批准了Adcetris®。
在一项开放单臂多中心临床试验中,评价Adcetris®对自身干细胞移植后复发HL患者的疗效。研究共纳入102例患者,主要终点是客观缓解率(ORR)和反应持续时间。结果显示,接受治疗的患者ORR为73%,反应持续时间的中位数为6.7个月。另一项开放单臂多中心临床II期试验中,评价Adcetris®对复发性sALCL患者的疗效,纳入患者58例,试验的主要终点是客观缓解率和反应持续时间。研究显示,接受治疗的患者ORR为86%,反应持续时间中位数为12.6个月。
03.Trastuzumab Emtansine(Kadcyla®:Genentech/Roche)
Kadcyla(Adptrastuzumab emtansine,恩美曲妥珠单抗)由罗氏和ImmunoGen共同研发,于2013年2月22日被FDA批准用于HER2阳性转移性乳腺癌。2020年1月21日,该药成功在中国获批。它是在我国首个上市的ADC药物,也是全球首款被批准用于实体瘤的ADC药物。2021年Kadcyla全球销售额为21.8亿美元,位居ADC药物销售的榜首。
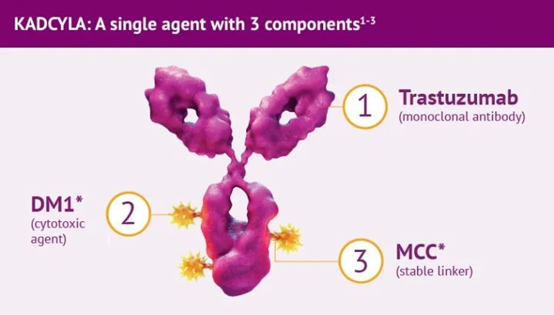
(Kadcyla®结构示意图,来源:罗氏)
Kadcyla属于第二代ADC,采用人源化单抗,更有效的细胞毒药物,即降低了免疫原性,又提升了毒性药物活性。它由微管抑制剂DM1通过不可清除的硫醚键连接子与靶向HER2的人源化IgG1偶联,经过受体介导的内化被溶酶体溶解,使DM1降解产物在肿瘤细胞内释放致使细胞凋亡。
Kadcyla的获批主要基于随机、开放标签、多中心临床III期KATHERINE研究的结果。
这项KATHERINE III期研究共纳入了1486例接受过赫赛汀和紫杉烷化疗等术前新辅助治疗的HER2阳性早期乳腺癌患者。患者在手术后12周内随机分组,分别接受T-DM1(3.6mg/kg,iv)或赫赛汀(6mg/kg,iv)维持治疗,每3w一次,持续14个周期。试验的主要终点是iDFS(无侵袭性疾病生存期),即从接受辅助治疗开始到浸润性乳腺癌复发或因任何原因死亡的时间。次要终点包括无病生存期(DFS)和总生存期(OS)。结果显示:T-DM1组的3年iDFS较赫赛汀组提高了11.3%,分别为88.3%vs 77.0%,复发风险降低了50%,差异非常显著(P<0.01)。亚组分析显示,无论激素受体状态,淋巴结状况,以及术前新辅助治疗中接受的HER2靶向治疗方案,都不影响T-DM1对iDFS的改善。
04.Inotuzumab Ozogamicin(Besponsa®:Pfizer/Wyeth)
Besponsa(Inotuzumab Ozogamicin)由辉瑞研发,2017年6月28日获得欧洲药物管理局(EMA)批准上市,2017年8月17日获美国FDA批准上市,2021年12月12日获得中国NMPA批准上市,用于治疗急性淋巴细胞白血病(ALL)。
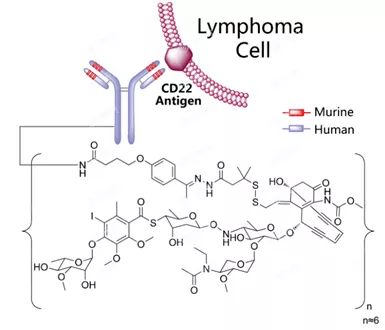
Inotuzumab Ozogamicin结构式
Besponsa是一种ADC药物,由靶向CD22的单克隆抗体inotuzumab与细胞毒制剂卡奇霉素(calicheamicin)偶联而成。CD22是在癌细胞上发现的一种细胞表面抗原,存在于几乎所有的B-ALL患者中。Besponsa靶向结合恶性B细胞表面的CD22抗原后内化进入细胞内,释放出细胞毒制剂卡奇霉素,摧毁癌细胞。
Besponsa的获批,是基于III期临床研究INO-VATE 1022的数据。
该研究是一项开放标签、随机研究,在326例复发或难治B细胞前体ALL成人患者中开展,评估了Besponsa相对于研究者选择的标准护理化疗方案的疗效和安全性。该研究有2个独立的主要终点:有或无血液学缓解的完全应答(CR/CRi),总生存期(OS)。数据显示,与标准护理化疗相比,Besponsa在多个评价指标方面均表现出改善,包括完全血液学缓解和无进展生存期(PFS)。
05.Moxetumomab pasudotox(Lumoxiti®:阿斯利康)
阿斯利康的Lumoxiti(Moxetumomab pasudotox,帕西妥莫单抗)于2018年9月13日获得FDA批准上市,用于治疗复发性或难治性毛细胞白血病(HCL,hairy cell leukemia)成人患者。这些患者至少接受过两种全身性治疗,其中包括嘌呤核苷类似物(PNA,purine nucleoside analog)疗法。Lumoxiti是过去20多年来,首个美国获批治疗HCL的药物。
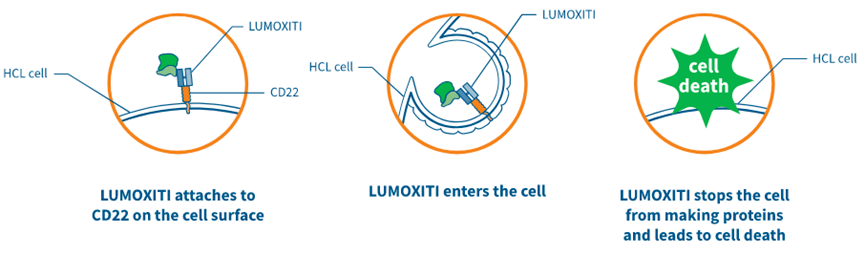
(Lumoxiti作用机制,来源:阿斯利康)
Lumoxiti的获批是基于一项III期临床研究(Study 1053)的数据。
该研究是一项单臂、多中心研究,在80例既往接受了至少2种方案的复发性或难治性HCL成人患者中开展,评估了Lumoxiti单药治疗的疗效和安全性。该研究在全球14个国家34个治疗中心开展。研究数据显示,Lumoxiti单药治疗的总缓解率为75%(95%CI:64-84),完全缓解率为41%(95%CI:30-53),持久的完全缓解率为30%(95%CI:20-41)。中位随访16.7个月后,中位完全缓解持续时间尚未达到。
06.Polatuzumab Vedotin(Polivy®:Genentech/Roche)
Polivy(polatuzumab vedotin,维博妥珠单抗)系由罗氏的基因泰克开发,2019年6月11日首次获得FDA加速批准上市,联合苯达莫司汀和利妥昔单抗,用于治疗至少两次以上的复发或难治性(R/R)弥漫性大B细胞淋巴瘤(DLBCL)的成年患者。
根据药融云中国药品审评数据显示,罗氏(Roche)已在中国提交了四项注射用维博妥珠单抗的上市申请,即将获批。
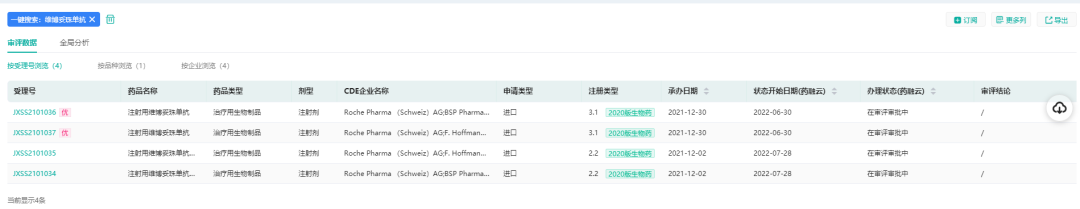
(数据来源:药融云中国药品审评数据)
Polivy首次批准基于一项开放标签、全球性多中心临床研究GO29365。
该研究的临床II期研究将80例过度预治疗的复发或难治性(R/R)弥漫性大B细胞淋巴瘤(DLBCL)患者随机分配至两个组:PBR组(polatuzumab vedotin+苯达莫司汀+利妥昔单抗)和BR组(苯达莫司汀+利妥昔单抗)。在治疗结束时,PBR组的完全缓解率(CR)为40%,BR组的CR为18%。PBR组最佳总体缓解率(完全和部分缓解)为63%,BR组则为25%。PBR组部分或完全缓解的25名患者中,16名(64%)的反应持续时间至少为6个月,12名(48%)的反应持续时间至少为12个月。
罗氏的这款ADC药物在2021年的全球销售额2.47亿瑞郎,已经在全球60多个国家或地区获批,市场潜力巨大。
07.Enfortumab Vedotin(Padcev®:Astellas Pharma/Seattle Genetics)
Padcev(Enfortumab Vedotin)由Seagen与安斯泰来研发,于2019年12月18日被FDA批准用于治疗晚期或转移性尿路上皮癌。Padcev是一种针对Nectin-4的ADC,Nectin-4是一种位于大多数尿路上皮癌细胞表面的黏附蛋白。它由一种完全的人IgG1-kappa抗体通过蛋白酶可裂解连接物与微管破坏剂MMAE结合而成。
Padcev的获批是基于全球III期临床试验EV-301。该实验纳入125名局部晚期或转移性的尿路上皮癌患者,这些患者既往都接受过PD-1/L1药物和铂类化疗。结果显示,44%的患者肿瘤显著缩小,其中12%的患者肿瘤完全消失。疗效持续的中位时间为7.6个月。
根据药融云中国临床试验最新数据显示,Padcev目前在国内处于III期临床。
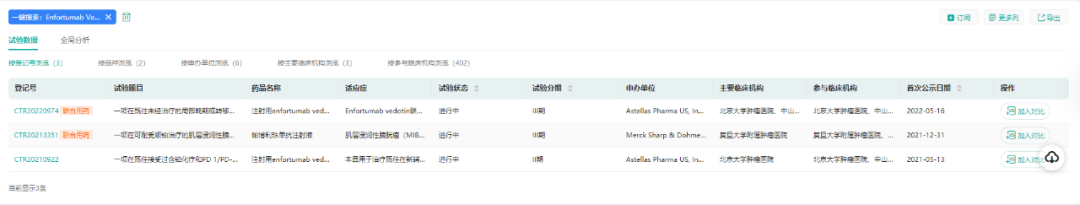
(数据来源:药融云中国临床试验)
08.Trastuzumab Deruxtecan(Enhertu®:Daiichi Sankyo/AstraZeneca)
Enhertu是阿斯利康和第一三共合作开发的靶向HER2的第三代ADC药物,于2021年12月20日被美国FDA批准上市,用于治疗既往已经接受过2种以上HER2靶向疗法的不可手术切除或转移性HER2阳性乳腺癌成人患者。2022年3月21日,Enhertu在中国的上市申请获得NMPA受理。
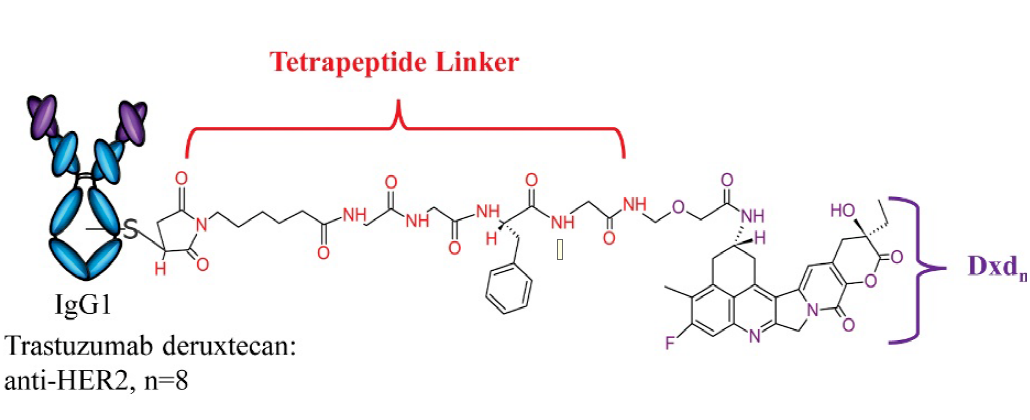
Trastuzumab Deruxtecan结构
Enhertu由第一三共独有的DXd技术平台设计开发,是第一三共肿瘤产品组合中的领先ADC药物,阿斯利康于2019年3月以69亿美元的免疫肿瘤学合作加入,共同开发Enhertu。因其优秀的临床试验结果数据,该药也被冠以神药之名。Enhertu(DS-8201)的综合临床研究成果,可谓是“实力抢镜”,虽然以后线治疗起家,但临床数据遍布乳腺癌、胃癌、肺癌、结直肠癌,给多癌种HER2突变患者带来了新的治疗希望,展现出非凡的潜力!
在2021年9月16~21日召开的欧洲肿瘤内科学会(ESMO)年会上,DS-8201多项研究结果再次刷新,横扫肺乳胃肠四大癌种,风采不减。
在DESTINY-Breast03的3期试验研究结果,这是首个头对头对比ADC药物(DS-8201和T-DM1)治疗HER2阳性晚期乳腺癌疗效与安全性的一项多中心、开放性、随机、Ⅲ期临床研究。纳入了先前在晚期或转移性环境中接受过曲妥珠单抗(赫赛汀)和紫杉类治疗的不可切除或转移性HER2阳性乳腺癌患者。值得注意的是,这些患者被允许具有临床稳定的、经治疗的脑转移。在2021年ESMO大会期间提交的试验数据显示,研究者评估的DS-8201的PFS为25.1个月(95%CI,22.1-NE),而T-DM1为7.2个月(95%CI,6.8-8.3)(HR,0.27);研究组或对照组均未达到中位生存期。上述数据表明,与TDM1相比,DS-8201能够降低HER2阳性转移性乳腺癌患者的疾病进展或死亡风险高达72%!
09.Sacituzumab Govitecan(Trodelvy®:Gilead/Immunomedics)
Trodelvy®由Immunomedics公司开发,2020年9月Gilead以210亿美元收购Immunomedics,获得了Trodelvy®的所有权。2020年4月22日,Trodelvy在美国上市,用于治疗先前已接受过至少两种疗法治疗的转移性三阴性乳腺癌成人患者,是全球首个获批的靶向TROP-2的ADC药物。
2022年6月7日,云顶新耀引进的注射用戈沙妥珠单抗(拓达维,Trodelvy)的上市申请已经获得批准,此次获批的适应症为:治疗接受过至少两种系统治疗(其中至少一种为针对转移性疾病的治疗)的不可切除的局部晚期或转移性三阴性乳腺癌成人患者。
2022年8月15日,云顶新耀发布公告,与吉利德子公司Immunomedics终止Trop2ADC药物Trodelvy的合作,Immunomedics向云顶新耀支付2.8亿美元预付款、1.75亿美元里程碑金额,协议总金额为4.55亿美元,约合30.67亿元人民币。
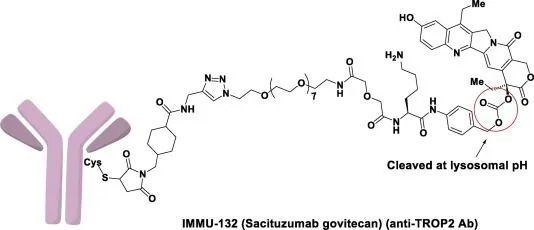
Trodelvy结构特征(图片来源:broadpharm.com)
2020年07月06日,Immunomedics宣布Trodelvy在治疗转移性三阴性乳腺癌(mTNBC)患者的确认性3期ASCENT临床试验中达到了无进展生存期(PFS)的主要终点,将患者疾病进展或死亡风险降低59%。
在ASCENT研究中,超过500名转移性三阴性乳腺癌患者随机接受Trodelvy或医生选择的化疗的治疗,这些患者至少接受过两种转移性疾病的既往疗法。试验结果表明:与化疗相比,Trodelvy将患者疾病进展或死亡风险降低59%(HR=0.41,95%CI,0.32-0.52)。Trodelvy治疗患者的中位PFS为5.6个月(95%CI,4.3-6.3),化疗为1.7个月(95%CI,1.5-2.6)(p<0.0001)。此外,Trodelvy还达到了研究的关键次要终点,包括总生存期和客观缓解率。
10.Belantamab Mafodotin(Blenrep®:GlaxoSmithKline)
2020年08月06号,葛兰素史克(GlaxoSmithKline,GSK)宣布,美国FDA已加速批准Blenrep(belantamab mafodotin-blmf)作为单药疗法,用于治疗既往接受过至少4种疗法(包括抗CD38单克隆抗体、蛋白酶体抑制剂和免疫调节剂)的复发性或难治性多发性骨髓瘤成人患者。
值得一提的是,Blenrep是全球获批的第一个BCMA靶向疗法,FDA加速批准Blenrep基于DREAMM-2试验的结果:ORR(31%)、DOR(73%的应答患者DOR≥6个月)。目前Blenrep在国内已经进行III期临床开发。
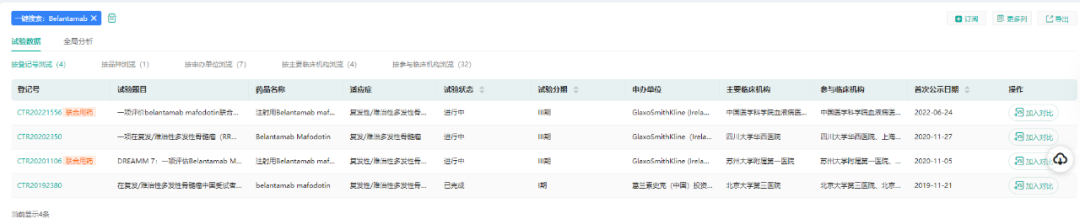
(数据来源:药融云中国临床试验)
2022年11月7日,GSK宣布在关于复发或难治性多发性骨髓瘤(RRMM)的III期试验DREAMM-3中,Blenrep未达到临床终点。
DREAMM-3研究是一项“头对头”的优效性试验:评估Blenrep单药与泊马度胺联合低剂量地塞米松(PomDex)的疗效对比。该项研究并未达到无进展生存期(PFS)这一主要临床终点。在该研究中,Blenrep的中位PFS为11.2月,PomDex为7月;次要终点包括总反应率(ORR)、缓解持续时间(DOR)和总生存期(OS)。Blenrep和PomDex的ORR分别为41%和36%;12个月的DOR率分别为76.8%和48.4%。此外,Blenrep的安全性与耐受性与先前一致,没有发现新的安全信号。
此次研究结果不佳,让该药发展前景变得不是非常乐观。2022年11月22日,葛兰素史克宣布应FDA要求做出将BCMA ADC药物Blenrep撤市的决定。
11.Cetuximab Saratolacan(Akalux®:Rakute Medical)
Akalux是一种靶向表皮生长因子受体(EGFR)的mAb,由西妥昔单抗(cetuximab)与IRDye700DX构成的抗体偶联药物(ADC),该药于2020年9月在日本获得了有条件批准用于治疗不可切除的局部晚期或复发性头颈癌。
由于添加了光反应物质,Akalux在与肿瘤靶向结合之后,可以在局部被由光导纤维释放的红色激光激活,从而导致肿瘤细胞的死亡。其独特之处在于,利用抗体介导的靶向递送实现高度肿瘤特异性,同时利用激光激活生物物理机制精确地诱导癌细胞的快速死亡,且避免伤害周围正常组织。
这是目前全球首次获批的头颈部肿瘤光免疫治疗药物,除了头颈癌,还有在EGFR过表达的食道癌、肺癌、结肠癌、胰腺癌等多种实体瘤应用。
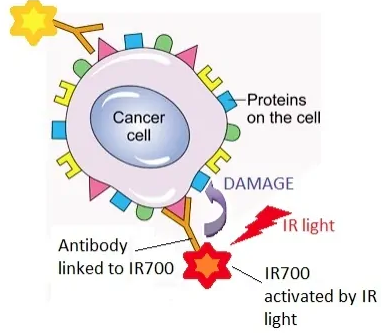
Bakalux作用机制
目前Akalux的临床试验主要集中在头颈癌,在一项针对不能通过手术、放疗或铂类化疗治疗的复发性头颈癌患者的IIa期临床研究中,共纳入30例患者,临床数据显示,客观缓解率(ORR)为28%(8/28),完全缓解率(CR)为14%(4/28)。28例可评估患者的中位无进展生存期(mPFS)为173天(5.7个月),全部30例患者的中位总生存时间(mOS)为278天(9.1个月)。研究结果显示,使用Akalux治疗安全且耐受性良好的。
12.loncastuximab tesirine(Zynlonta®:ADC Therapeutics)
ADC Therapeutics公司2021年4月23日宣布,美国食品和药物管理局(FDA)已批准Zynlonta(loncastuximab tesirine-lpyl),用于治疗已接受过2种或多种系统疗法的复发或难治性(r/r)大B细胞淋巴瘤(LBCL)成人患者,包括弥漫性大B细胞淋巴瘤(DLBCL)、起源于低级别淋巴瘤和高级别细胞淋巴瘤的DLBCL。
该药目前在国内已经进行到III期临床,临床进展正在平稳推进。值得一提的是,Zynlonta是第一个也是目前唯一一个CD19靶向ADC药物。

(数据来源:药融云中国临床)
FDA的加速批准基于总缓解率数据,针对该适应症的继续批准将取决于验证性试验中对临床益处的验证和描述。
来自关键LOTIS 2试验的数据显示,在过度预治疗(heavily-preteated,指先前接受过多种疗法)r/r DLBCL患者中,Zynlonta单药治疗的总缓解率(ORR)达到了48.3%、完全缓解率(CR)为24.1%,并且缓解持久。(详见:Zynlonta FDA Approval Presentation)。
13.维迪西妥单抗(爱地希®:荣昌生物)
2021年6月9日,中国国家药监局(NMPA)宣布,已通过优先审评审批程序附条件批准注射用维迪西妥单抗(商品名:爱地希)上市,适用于至少接受过2种系统化疗的HER2过表达局部晚期或转移性胃癌(包括胃食管结合部腺癌)患者的治疗。
一项2期注册性临床试验数据显示,维迪西妥单抗治疗上述适应症患者,客观缓解率(ORR)为24.4%,无进展生存期(PFS)中位数为4.1个月,总生存期(OS)中位数为7.6个月。
2021年7月初,维迪西妥单抗正式全国开售。药融云数据显示,本品价格进入医保谈判前价格为60mg/支/盒对应13500元/盒。进入医保目录后,为3800元/盒。在2022年JPM大会上,荣昌生物表示:本款HER2-ADC的销售部团队目前有200人,已覆盖30个省份130个城市的400家医院和405家药店。
2022年1月5日,荣昌生物宣布:注射用维迪西妥单抗(商品名:爱地希)新适应症获得国家药品监督管理局(NMPA)的上市许可批准,用于治疗既往接受过含铂化疗且HER2过表达即免疫组化检查结果为2+或3+的局部晚期或转移性尿路上皮癌患者。该适应症于2021年7月14日提交上市申请获受理,耗时5个月迅速获批,成为国内首个靶向HER2治疗尿路上皮癌的ADC药物。

流行病学数据显示,尿路上皮癌是常见恶性肿瘤之一,发病率及死亡率均占男性泌尿生殖系统肿瘤的首位,九成起源于膀胱,也可见于肾盂、输尿管。据沙利文报告,全球新增尿路上皮癌病例于2030年将达到约66.2万例,2025年至2030年复合年增长率为2.5%;在中国,尿路上皮癌的发病率增速高于全球水平,预计于2030年将达到约10.6万例。统计显示,该病复发率和转移率较高,约20%的患者确诊时已发生转移或病程已进展至不可切除阶段,现有治疗手段远未满足巨大的临床需求。
值得注意的是,荣昌生物在2021年8月与国际知名生物制药公司西雅图基因(Seagen Inc.纳斯达克:SGEN)达成一项全球独家许可协议:针对荣昌生物的ADC药物维迪西妥单抗(RC48,商品名:爱地希),Seagen将付出2亿美元首付款+最高24亿美元的里程碑付款,以获得该药物全球部分地区的开发和商业化权益。目前已经收到了2亿美元首付款。
14.tisotumab vedotin-tftv(Tivdak®:Genmab/Seagen)
2021年9月20日,美国FDA加速批准ADC药物(抗体偶联)Tivdak(tisotumab vedotin-tftv),用于治疗在化疗期间或化疗后病情进展的复发性或转移性宫颈癌成人患者。该加速批准基于肿瘤缓解和缓解持久性数据,针对该适应症的后续完全批准,将取决于确证性临床试验中对临床益处的验证和描述。
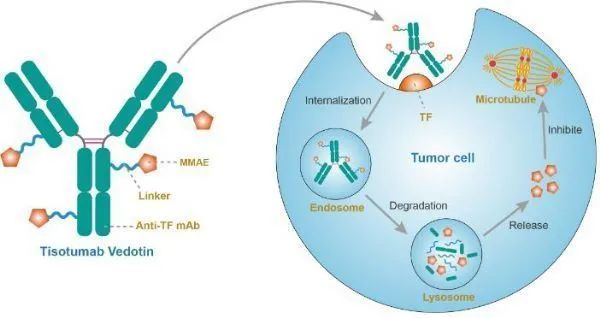
tisotumab vedotin作用机制(图片来源:immunopaedia.org.za)
此次批准基于关键II期innovaTV 204研究的结果。相关数据已于2020年9月在欧洲医学肿瘤学会(ESMO)虚拟大会上公布。结果显示,Tivdak作为一种单药疗法具有显著疗效,可提供具有临床意义和持久的客观缓解:总缓解率(ORR)为24%、中位缓解持续时间(DOR)为8.3个月,且安全性可控。Tivdak的批准上市,对于复发性或转移性宫颈癌女性患者来说是一个重大进步。
15.mirvetuximab soravtansine(Elahere®:ImmunoGen/华东医药)
2022年11月14日,Elahere于美国获加速批准上市,用于治疗叶酸受体α(FRα)阳性且既往接受过1-3线全身治疗方案的铂耐药卵巢上皮性癌、输卵管癌或原发腹膜癌的成年患者,Elahere也由此成为美国FDA批准的首个用于铂耐药卵巢癌的ADC药物。
华东医药拥有Elahere在大中华区(含中国大陆、香港、澳门和台湾地区)的独家临床开发及商业化权益,目前该药在国内处于III期临床,公司计划于2023年下半年向NMPA递交BLA申请。
(数据来源:药融云中国临床试验)
此次美国FDA的加速批准是基于一项关键性III期临床试验SORAYA的结果。在该试验中,主要评估了Elahere治疗FRα阳性、铂耐药卵巢癌患者的有效性和安全性。
该试验的可评估疗效的人群包括104例铂耐药患者。入组患者的人群特征包括:中位年龄为62岁;96%的患者为白人,2%的患者为亚洲人;ECOG评分为0(57%)或1(43%);10%的患者之前接受过1个系统治疗线,39%的患者之前接受过2个系统治疗线,50%的患者之前接受过3个系统治疗线;所有患者之前都接受过贝伐珠单抗治疗,47%的患者之前接受过PARP抑制剂。
该试验的主要终点为研究者评估的客观缓解率(ORR);关键的次要终点是根据RECIST v1.1标准评估的中位持续缓解时间(DOR)。该试验结果显示,已达到主要终点,经研究者评估确认的客观缓解率(ORR)为31.7%(95%,置信区间:22.9%~41.6%),其中,完全缓解(CR)率为4.8%,部分缓解(PR)率为26.9%。截至2022年3月3日,研究者评估的中位持续缓解时间(DOR)为6.9个月(95%,置信区间:5.6~8.1)。
Elahere于美国获加速批准上市,对其在中国获批上市构成积极影响,对华东医药的ADC药物发展也有深远影响。
参考来源:
[1] 药融云数据库
[2] 各公司公开信息
想要解锁更多药品信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药品各国上市情况、药品批文信息、销售情况与各维度分析、市场竞争格局、药企申报审批信息、最新动态与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!

—END—





收藏
登录后参与评论