





创新药研发是通过疾病靶点研究、结构筛选、工艺设计与优化(或处方与工艺设计)、结构确证、质量控制、稳定性研究、药效与药代研究、安全性评价(毒理研究)等一系列研究,将一个全新分子投入到市场用于治疗特定疾病的复杂过程。这个过程中,安全性评价是不可或缺的重要环节,也是新药研发终止的重要原因之一,是创新药研发永恒关注的问题。
临床前安全性评价试验样品包括原料药安全性评价样品及制剂安全性评价样品,分别评价新化学实体及新制剂的安全性。
临床前安全性评价主要是动物毒理试验,是创新药从动物研究到人体研究的桥梁,通过动物毒理试验①可以初步判定药物的毒副作用,为开展临床提供安全性数据支持。②可以为杂质限度制定提供依据;③依据试验结果,在安全性可支持的范围内杂质水平可适当放宽,可以一定程度上降低临床试验样品的生产压力,不必花费大量时间过度优化工艺,为项目推进争取时间;④如在毒理试验中发现不可接受的毒性可及时止损;⑤在后期临床研究中如出现不良反应,临床前毒理数据可为原因调查提供依据;⑥毒理试验结果可为制剂开发规格制定提供重要支撑。
安全评价样品杂质谱的控制至关重要。正常情况下,产品工艺开发与优化过程是追求杂质最小化的过程,样品纯度越高,安全性越好;而在毒理研究中,样品纯度不是追求的目标,因为毒理样品中的杂质肩负着杂质安全性评价的重任,只有存在于毒理样品中才能获得评价,积累安全性数据。
(1)有关物质
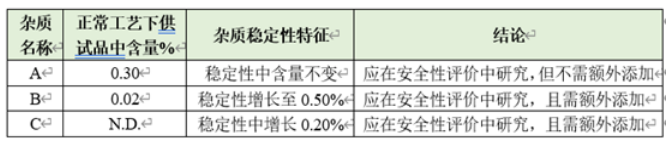
l在稳定工艺下,多批样品中均存在,且原料药中含量超过0.1%(或制剂中含量超过0.15%)的杂质(工艺杂质),如下表中杂质A。
l样品中存在,含量较低,但在稳定性考察中含量显著增长的杂质(工艺杂质&降解杂质),如下表中杂质B。
l样品中不存在,但在稳定性考察中新增产生的杂质(降解杂质),如下表中杂质C。
(2)异构体
异构体与API结构相同,但药效、药代、毒性可能具有显著差异,是产生毒副作用风险来源之一,FDA要求对手性药物进行毒理研究时,应获得各单一构型的立体异构体,进行必要的比较研究,以确定拟进一步开发的药物。从这一条规定看,立体异构体在确定候选药物的时候已需毒理研究,理论上在临床前安全性评价时,可以不再重复考察,但为进一步确认异构体的安全限度,可以选用异构体含量稍高的样品或添加适当浓度异构体进行安全性评价研究。
(3)遗传毒杂质
终产品中遗传毒杂质一定会被严格控制在安全限度之下,不会产生难以预料或不可接受的毒性,因此临床前安全性评价样品中通常不需要额外添加遗传毒杂质。如确实需考察其安全性应通过定向合成单独考察其毒性。
(4)聚合物
聚合物与有关物质的研究策略基本一致,如果聚合物含量较高或稳定性中明显增长的话需要额外添加考察其安全性。
无机杂质
通常拥有充分的安全性研究资料并具有确定的限度(ICH Q3D),一般无需额外添加考察安全性。
残留溶剂
通常拥有充分的安全性研究资料并具有确定的限度(ICH Q3C),一般无需额外添加考察安全性。

安全性评价样品中杂质水平是另一个需要重点考察的因素,也是一个难度系数超高的技术活。杂质水平过高,可能影响API毒理评价结果,造成安全性评价试验失败;杂质水平过低,其所能支持的杂质安全水平有限,可能给后面临床批样品生产造成压力,耽误研发进程。其中的风险与获益需要仔细评估。
杂质添加水平是没有硬性规定的,个人认为杂质水平可以根据早期有限的毒性研究数据,结合目前的工艺水平和稳定性来制定,也就是根据预期的毒理给药水平、目前工艺下样品中杂质的水平及稳定性,倒推出毒理样品中的杂质水平。例如,基于早期研究预期大鼠长毒Noael不会低于20 mg/kg/d,临床拟定的给药剂量为120 mg/d,目前工艺下样品中杂质含量为0.6%,那么倒推出安全性评价样品中该杂质的水平应为=[(120 mg/kg/d 60 kg)0.6%37 kg/m2]/(20 mg/kg/d6 kg/m2)=0.37%。

CDE要求临床批样品中杂质水平不得超出动物安全性试验数据所支持的相应杂质的水平。那么,安全性评价样品所能支持的水平怎样计算呢?尤其是早期的IND阶段可以怎样计算呢?
计算的方式有很多种,核心原理是动物Noael所代表的暴露量的与人体之间的转化。下面借鉴了文献中的两种计算方式:


这里需要说明的是,以上的这些依据安全性评价结果推算杂质限度的方法仅适用于研发初期,因为此时原料药、制剂工艺均尚未完全确定,杂质限度可根据安全性评价结果制定,只要确保临床试验安全即可,到了研发中后期,原料药、制剂工艺均尚基本确定,杂质限度原则上应符合ICH要求。

安全性评价用样品的制备方法有以下几种:
(1)杂质可获得情况下
- 选择一批杂质种类相对较全的原料药产品,向其中添加一定水平的需要重点评估的杂质(因为是固体混合,因此要特别注意混合均匀性)。
- 在反应过程中加入一定水平的杂质(需要提前考察各杂质在反应中的清楚和稳定性)
(2)杂质不可获得的情况下
- 对正常生产所获得的产品进行适当的降解破坏,获得所需的杂质水平
- 通过调整工艺中某些参数获得较高杂质水平的样品。

安全性评价样品在什么时候提供比较适宜呢?首先看下安全性评价试验的特点和审评要求:
①临床前安全性评价试验时间跨度较长;
②部分研究需得到安全性评价结果(或进行中的部分结果)的支持才能开展;
③临床前安全性评价试验是IND申报的审评重点,直接关系是否可进一步开展临床研究,故CDE要求必须完成临床前安全性评价才能申报。
综合以上几点,在实际研究过程中,安全性评价试验常常是申报的限速步骤,因此在工艺确认之后应尽快生产安全性评价样品,尽早开展安全性评价试验(见下图)。


理论上用于安全评价用样品的检测方法最好是基本确认的方法,方便杂质的比对,否则,如果检测方法后期发生较大改动,需要桥接两个方法,将杂质一一比对,不要造成杂质谱分析混淆。

应对临床前安全性评价样品进行稳定性考察,对影响安全性、有效性的重要指标进行检测,如外观、杂质水平、主成分含量、晶型等。对临床前安全性评价样品进行稳定性研究,一是要确保安全性评价期间样品质量可控,二是为临床前安全性评价样品的运输和储存条件的选择提供依据。

在新药研发中,安全性评价样品是较为特殊和重要的存在,作为新药研发人员,我们应充分理解非临床安全性评价的相关要求,充分掌握非临床安全性评价对样品的质量要求;充分利用非临床安全性评价结果在质量研究中的支撑作用,助力新药更快、更稳向前推进。
参考文献
[1] 化学药物杂质研究技术指导原则
[2] 黄晓龙,对创新药研发中杂质限度确定的几点思考,中国新药杂质 2007,16(2),97-100。
[3]何伍,王海学,浅谈药物杂质限度的制订方法,中国医药工业杂志,2009,40(10),787-790。
[4]手性药物研究指导原则
<END>
要解锁更多企业药品研发信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药品各国上市情况、药品批文信息、销售情况与各维度分析、市场竞争格局、一致性评价情况、集采中标情况、药企申报审批信息、最新动态与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!






收藏
登录后参与评论